
TALZENNA 0,25 mg, gélule, boîte de 1 flacon de 30
Dernière révision : 30/08/2024
Taux de TVA : 2.1%
Prix de vente : 1 273,70 €
Taux remboursement SS : 100%
Base remboursement SS : 1 273,70 €
Laboratoire exploitant : PFIZER
Source :

Cancer du sein
Talzenna est indiqué en monothérapie pour le traitement des patients adultes atteints d'un cancer du sein localement avancé ou métastatique HER2 négatif et présentant des mutations germinales BRCA1/2. Les patients doivent avoir été précédemment traités par une anthracycline et/ou un taxane au stade (néo)adjuvant, localement avancé ou métastatique, sauf s'ils n'étaient pas éligibles à ce type de traitement (voir rubrique Propriétés pharmacodynamiques). Les patients atteints d'un cancer du sein positif aux récepteurs hormonaux (RH) doivent préalablement avoir reçu une hormonothérapie ou être considérés comme non-éligibles à une hormonothérapie.
Cancer de la prostate
Talzenna est indiqué en association avec l'enzalutamide pour le traitement des patients adultes atteints d'un cancer de la prostate métastatique résistant à la castration (CPRCm) pour lesquels la chimiothérapie n'est pas cliniquement indiquée.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Allaitement (voir rubrique Fertilité, grossesse et allaitement).
Myélosuppression
Des cas de myélosuppression, incluant anémie, leucopénie/neutropénie et/ou thrombocytopénie, ont été rapportés chez des patients traités par talazoparib (voir rubrique Effets indésirables). Le traitement par talazoparib ne doit pas être débuté tant que le patient n'a pas récupéré d'une toxicité hématologique induite par une thérapie antérieure (≤ grade 1).
Des précautions doivent être prises en vue de surveiller régulièrement les paramètres hématologiques et les signes et symptômes associés à l'anémie, la leucopénie/neutropénie et/ou la thrombocytopénie chez les patients recevant du talazoparib. En cas de survenue de tels événements, des modifications de dose (réduction ou interruption) sont recommandées (voir rubrique Posologie et mode d'administration). Une prise en charge symptomatique, avec ou sans transfusion de sang et/ou de plaquettes et/ou administration de facteurs de croissance granulocytaires peut être instaurée en fonction des besoins.
Syndrome myélodysplasique/leucémie aiguë myéloïde
Des cas de syndrome myélodysplasique/leucémie aiguë myéloïde (SMD/LAM) ont été rapportés chez des patients traités par des inhibiteurs de la poly (adénosine diphosphate-ribose) polymérase (PARP), y compris le talazoparib. Dans l'ensemble, des cas de SMD/LAM ont été rapportés chez < 1 % des patients atteints de tumeurs solides traités par talazoparib dans les études cliniques (voir rubrique Effets indésirables). Les facteurs susceptibles de contribuer au développement d'un SMD/LAM incluent des antécédents de chimiothérapie à base de platine, d'autres agents endommageant l'ADN ou de radiothérapie. L'hémogramme doit être obtenu avant le début du traitement par talazoparib et surveillé chaque mois afin d'identifier des signes éventuels de toxicité hématologique pendant le traitement. Si un SMD/LAM est confirmé, le traitement par talazoparib doit être arrêté.
Événements thromboemboliques veineux
Chez les patients atteints de CPRCm, une incidence plus élevée d'événements thromboemboliques veineux a été observée avec Talzenna en association avec l'enzalutamide par rapport à l'enzalutamideseul. Les patients doivent être surveillés afin de détecter tout signe clinique ou symptôme de thrombose veineuse profonde et d'embolie pulmonaire et doivent recevoir les soins médicaux appropriés (voir rubrique Effets indésirables).
Contraception chez les femmes en âge de procréer
Le talazoparib s'est révélé clastogène dans un test d'aberrations chromosomiques in vitro sur des lymphocytes du sang périphérique humain et dans un test des micronoyaux sur moelle osseuse in vivo chez le rat, mais non mutagène dans un test d'Ames (voir rubrique Données de sécurité préclinique), et est susceptible d'avoir des effets délétères sur le fœtus lorsqu'il est administré à une femme enceinte. Les femmes enceintes doivent être informées du risque potentiel pour le fœtus (voir rubrique Fertilité, grossesse et allaitement). Les femmes en âge de procréer ne doivent pas démarrer une grossesse pendant le traitement par Talzenna et ne doivent pas être enceintes au début du traitement. Toutes les femmes en âge de procréer doivent faire un test de grossesse avant le traitement.
Les femmes doivent utiliser une méthode de contraception hautement efficace pendant le traitement par Talzenna et pendant au moins 7 mois après la fin du traitement. Etant donné que la contraception hormonale n'est pas recommandée chez les patientes atteintes d'un cancer du sein, deux méthodes de contraception non hormonales et complémentaires doivent être utilisées (voir rubrique Fertilité, grossesse et allaitement).
Il doit être recommandé aux patients de sexe masculin ayant des partenaires féminines en âge de procréer ou des partenaires enceintes d'utiliser une méthode de contraception efficace (même après une vasectomie) pendant le traitement par Talzenna et pendant au moins 4 mois après la dernière dose.
Résumé du profil de sécurité
Le profil de sécurité global de Talzenna repose sur les données combinées de 1 088 patients, dont 690 ayant reçu 1 mg de talazoparib par jour en monothérapie dans des études cliniques portant sur des tumeurs solides et 398 atteints de CPRCm ayant reçu le talazoparib 0,5 mg en association avec l'enzalutamide 160 mg dans le cadre de l'étude TALAPRO-2.
Les effets indésirables les plus fréquents (= 20 %) chez les patients recevant le talazoparib dans ces études cliniques étaient : anémie (55,6 %), fatigue (52,5 %), nausées (35,8 %), neutropénie (30,3 %), thrombocytopénie (25,2 %) et la perte d'appétit (21,1 %). Les effets indésirables de grade = 3 les plus fréquents (= 10 %) du talazoparib étaient : anémie (39,2 %), neutropénie (16,5 %) et thrombocytopénie (11,1 %).
Des modifications de la dose (réductions ou interruptions) en raison d'un effet indésirable sont survenues chez 58,7 % des patients recevant Talzenna 1 mg en monothérapie. Les effets indésirables les plus fréquents conduisant à des modifications de la dose étaient l'anémie (33,5 %), la neutropénie (11,7 %) et la thrombocytopénie (9,9 %).
Un arrêt définitif en raison d'un effet indésirable est survenu chez 2,9 % des patients recevant Talzenna, le plus fréquent étant l'anémie (0,6 %). La durée médiane d'exposition était de 5,6 mois (intervalle de 0,0 à 70,2).
Des interruptions du traitement Talzenna en raison d'effets indésirables sont survenues chez 62,1 % des patients atteints de CPRCm recevant Talzenna en association avec l'enzalutamide ; le plus fréquent étant l'anémie (44 %). Des réductions de posologie de Talzenna en raison d'effets indésirables sont survenues chez 52,8 % des patients, le plus fréquent étant l'anémie (43,2 %). Un arrêt définitif de Talzenna en raison d'effets indésirables est survenu chez 18,8 % des patients ; le plus fréquent étant l'anémie (8,3 %). La durée médiane d'exposition au talazoparib était de 86 semaines (intervalle de 0,29 à 186,14).
Liste des effets indésirables sous forme de tableau
Le tableau 4 récapitule les effets indésirables sur la base des données combinées listés par classe de systèmes d'organes et par catégorie de fréquence. Les catégories de fréquence sont définies de la façon suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10) et peu fréquent (≥ 1/1 000 à < 1/100). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 4. Effets indésirables sur la base des données combinées issues de 8 études (N = 1 088)
|
Classe de systèmes d'organes Fréquence Terme préférentiel |
Tous grades n (%) |
Grade 3 n (%) |
Grade 4 n (%) |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) Peu fréquent Syndrome myélodysplasique/leucémie aiguë myéloïdea |
2 (0,2) |
1 (< 0,1) |
1 (< 0,1) |
|
Affections hématologiques et du système lymphatique Très fréquent Thrombocytopénieb Anémiec Neutropénied Leucopéniee Fréquent Lymphopénief |
274 (25,2) 605 (55,6) 330 (30,3) 195 (17,9) 88 (8,1) |
88 (8,1) 411 (37,8) 163 (15,0) 52 (4,8) 37 (3,4) |
33 (3,0) 16 (1,5) 17 (1,6) 2 (0,2) 4 (0,4) |
|
Troubles du métabolisme et de la nutrition Très fréquent Diminution de l'appétit |
230 (21,1) |
11 (1,0) |
0 (0,0) |
|
Affections du système nerveux Très fréquent Sensations vertigineuses Céphalées Fréquent Dysgueusie |
157 (14,4) 207 (19,0) 68 (6,3) |
4 (0,4) 8 (0,7) 0 (0,0) |
1 (< 0,1) NA 0 (0,0) |
|
Affections vasculaires Fréquent Thromboembolie veineuse*g |
36 (3,3 %) |
23 (2,1 %) |
2 (0,2 %) |
|
Affections gastro-intestinales Très fréquent Vomissements Diarrhée Nausées Douleurs abdominalesh Fréquent Stomatite Dyspepsie |
167 (15,3) 205 (18,8) 389 (35,8) 162 (14,9) 54 (5,0) 69 (6,3) |
9 (0,8) 4 (0,4) 10 (0,9) 12 (1,1) 0 (0,0) 0 (0,0) |
0 (0,0) 0 (0,0) NA NA 0 (0,0) NA |
|
Affections de la peau et du tissu sous-cutané Très fréquent Alopécie |
189 (17,4) |
NA |
NA |
|
Troubles généraux et anomalies au site d'administration
Très fréquent Fatiguei |
571 (52,5) |
58 (5,3) |
NA |
Abréviations : n = nombre de patients ; NA = non applicable.
* Des effets indésirables de grade 5 ont été rapportés.
a. Voir également rubrique Mises en garde spéciales et précautions d'emploi.
b. Inclut les termes préférentiels thrombocytopénie et diminution de la numération plaquettaire.
c. Inclut les termes préférentiels anémie, diminution de l'hématocrite, diminution de l'hémoglobine et diminution du nombre des globules rouges.
d. Inclut les termes préférentiels neutropénie et diminution du nombre de neutrophiles.
e. Inclut les termes préférentiels leucopénie et diminution du nombre de globules blancs.
f. Inclut les termes préférentiels diminution du nombre de lymphocytes et lymphopénie.
g Inclut les termes préférentiels embolie pulmonaire, thrombose veineuse profonde, embolie veineuse et thrombose veineuse. Voir également rubrique Mises en garde spéciales et précautions d'emploi.
h. Inclut les termes préférentiels douleur abdominale, douleur abdominale haute, gêne abdominale et douleur abdominale basse.
i. Inclut les termes préférentiels fatigue et asthénie.
Description d'effets indésirables sélectionnés
Myélosuppression
Des effets indésirables liés à une myélosuppression de type anémie, neutropénie et thrombocytopénie ont été très fréquemment rapportés chez les patients traités par talazoparib. Des événements liés à une myélosuppression de grade 3 et de grade 4 ont été rapportés pour l'anémie (37,8 % et 1,5 % des patients, respectivement), la neutropénie (15,0 % et 1,6 % des patients, respectivement) et la thrombocytopénie (8,1 % et 3,0 % des patients, respectivement). Aucun décès n'a été rapporté en raison d'effets indésirables liés à une myélosuppression.
Dans les études en monothérapie (patients traités par 1 mg/jour), les événements indésirables liés à une myélosuppression les plus fréquents étaient l'anémie (33,5 %), la neutropénie (11,7 %) et la thrombocytopénie (9,9 %) rapportés chez environ 30 % des patients traités par 1 mg/jour de talazoparib et l'effet indésirable associé à l'arrêt définitif du traitement à l'essai était l'anémie, rapportée chez 0,6 % des patients.
Chez les patients atteints de CPRCm traités par talazoparib en association avec l'enzalutamide, l'anémie a entraîné l'interruption du talazoparib chez 44,0 % des patients, la diminution du nombre de neutrophiles chez 13,6 % et la diminution du nombre de plaquettes chez 7,8 %. Dans l'ensemble, 42,5 % des patients ont eu besoin d'une transfusion sanguine. La transfusion sanguine la plus fréquente était celle de concentrés de globules rouges (39,2 %). Un arrêt du traitement en raison d'une anémie, d'une neutropénie et d'une thrombocytopénie est survenu, respectivement, chez 8,3 %, 3,3 % et 0,5 % des patients.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT
traitement
- Sélection
des patients :
Cancer du
sein : les patients doivent être sélectionnés pour le traitement du cancer
du sein par Talzenna sur la base dela présence de mutations germinales BRCA
délétères ou soupçonnées de l'être, déterminées par un laboratoire expérimenté
utilisant une méthode de test validée. Le conseil génétique pour les patients
présentant des mutations BRCA doit être effectué, le cas échéant, conformément
aux réglementations locales.
Cancer de la
prostate : il n'est pas nécessaire de procéder à une recherche
mutationnelle pour sélectionner les patients atteints de CPRCm en vue d'un
traitement par Talzenna.
- Toutes les femmes en âge de procréer doivent effectuer un
test de grossesse avant le traitement.
SURVEILLANCE pendant le traitement : hémogramme avant instauration du traitement puis
tous les mois et si cliniquement indiqué.
L'utilisation de talazoparib en association avec l'enzalutamide n'est pas recommandée chez les patients présentant une insuffisance hépatique sévère (classe C sur l'échelle de Child-Pugh) car la pharmacocinétique et la sécurité n'ont pas été établies chez ces patients.
- FEMME en âge de PROCREER : utiliser une méthode de contraception hautement efficace avant le début du traitement, pendant le traitement et pendant les 7 mois suivant son arrêt. Etant donné que la contraception hormonale n'est pas recommandée chez les patientes atteintes d'un cancer du sein, deux méthodes de contraception non hormonales et complémentaires doivent être utilisées.
- HOMME dont la partenaire féminine est en âge de procréer ou enceintes : utiliser une méthode de contraception efficace (même après une vasectomie) pendant le traitement et pendant au moins 4 mois après la dernière dose.
- NE PAS ALLAITER au cours du traitement et pendant au moins 1 mois après la dernière dose.
- EVITER la curcumine (ingrédient courant du curry) pendant le traitement.
- PRUDENCE en cas de consommation de millepertuis (Hypericum perforatum) pendant le traitement
- INFORMER IMMEDIATEMENT le médecin en cas de :
- . Essoufflement, fatigue importante, pâleur ou battements cardiaques rapides.
- . Infection, frissons, fièvre ou sensation de chaleur.
- . Bleus ou saignements durant plus longtemps que d'habitude en cas de blessure.
- . Douleur ou raideur, gonflement et rougeur de la jambe (ou du bras) concernée, douleur thoracique, étourdissement.
- PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (fatigue, asthénie, sensations vertigineuses).
Femmes en âge de procréer/Contraception chez les hommes et les femmes
Les femmes en âge de procréer ne doivent pas démarrer une grossesse pendant le traitement par Talzenna et ne doivent pas être enceintes au début du traitement. Toutes les femmes en âge de procréer doivent effectuer un test de grossesse avant le traitement (voir rubrique Mises en garde spéciales et précautions d'emploi).
Les femmes en âge de procréer doivent utiliser des méthodes de contraception hautement efficaces (voir rubrique Mises en garde spéciales et précautions d'emploi) avant le début du traitement par talazoparib, pendant le traitement et pendant les 7 mois suivant l'arrêt du traitement par talazoparib. Etant donné que la contraception hormonale n'est pas recommandée chez les patientes atteintes d'un cancer du sein, deux méthodes de contraception non hormonales et complémentaires doivent être utilisées. Il doit être recommandé aux patients de sexe masculin ayant des partenaires féminines en âge de procréer ou des partenaires enceintes d'utiliser une méthode de contraception efficace (même après une vasectomie) pendant le traitement par Talzenna et pendant au moins 4 mois après la dernière dose (voir rubrique Mises en garde spéciales et précautions d'emploi).
Grossesse
Il n'existe pas de données sur l'utilisation de Talzenna chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité embryofœtale (voir rubrique Données de sécurité préclinique). Talzenna est susceptible d'avoir des effets délétères pour le fœtus en cas d'utilisation chez une femme enceinte. Talzenna n'est pas recommandé pendant la grossesse ou chez les femmes en âge de procréer n'utilisant pas de contraception (voir rubrique Mises en garde spéciales et précautions d'emploi).
Allaitement
On ne sait pas si le talazoparib est excrété dans le lait maternel. Un risque pour les enfants nourris au lait maternel ne peut être exclu. Par conséquent, l'allaitement est contre-indiqué (voir rubrique Contre-indications) pendant le traitement par Talzenna et pendant au moins 1 mois après la dernière dose.
Fertilité
Aucune information concernant la fertilité chez les patients n'est disponible. Au vu des effets non cliniques observés sur les testicules (partiellement réversibles) et les ovaires (réversibles), la fertilité masculine pourrait être compromise par le traitement par Talzenna (voir rubrique Données de sécurité préclinique).
Le talazoparib est un substrat des transporteurs de médicaments P-gp et protéine de résistance au cancer du sein (BCRP) et il est éliminé principalement par clairance rénale sous forme inchangée.
Agents pouvant affecter la concentration plasmatique de talazoparib
Inhibiteurs de la P-gp
Effet de l'enzalutamide
La co-administration de 160 mg d'enzalutamide augmente d'environ 2 fois l'exposition au talazoparib. L'administration de talazoparib 0,5 mg par jour en association avec l'enzalutamide permet d'obtenir approximativement la même concentration résiduelle (Crésid) à l'état d'équilibre que celle rapportée pour le talazoparib 1 mg par jour (voir rubrique Propriétés pharmacocinétiques). Lorsque Talzenna est co-administré avec l'enzalutamide, la dose initiale de Talzenna est de 0,5 mg (voir rubrique Posologie et mode d'administration). L'effet d'interaction sur le talazoparib d'autres dosages que 160 mg d'enzalutamide n'a pas été quantifié.
L'effet de la co-administration d'autres inhibiteurs de la P-gp sur l'exposition au talazoparib lorsque celui-ci est administré en association avec l'enzalutamide n'a pas été étudié. Si la co-administration d'inhibiteurs de la P-gp ne peut être évitée, lorsque Talzenna est administré avec l'enzalutamide, le patient doit être surveillé afin de déceler tout effet indésirable accru potentiel.
Effet d'autres inhibiteurs de la P-gp
Les données provenant d'une étude sur les interactions médicamenteuses chez des patients atteints de tumeurs solides à un stade avancé ont indiqué que l'administration concomitante de plusieurs doses quotidiennes d'un inhibiteur de la P-gp, l'itraconazole, à raison de 100 mg deux fois par jour avec une dose unique de 0,5 mg de talazoparib a augmenté l'exposition totale (AUCinf) et la concentration maximale (Cmax) du talazoparib de respectivement 56 % et 40 % environ, comparativement à une dose unique de 0,5 mg de talazoparib administrée seule. Une analyse de pharmacocinétique (PK) de population a par ailleurs montré que l'utilisation concomitante d'inhibiteurs puissants de la P-gp augmentait l'exposition au talazoparib de 45 %, par rapport au talazoparib seul.
L'utilisation concomitante d'inhibiteurs puissants de la P-gp (incluant mais ne se limitant pas à amiodarone, carvédilol, clarithromycine, cobicistat, darunavir, dronédarone, érythromycine, indinavir, itraconazole, kétoconazole, lapatinib, lopinavir, propafénone, quinidine, ranolazine, ritonavir, saquinavir, télaprévir, tipranavir et vérapamil) doit être évitée. Si l'administration concomitante avec un inhibiteur puissant de la P-gp est inévitable, la dose de Talzenna doit être réduite (voir rubrique Posologie et mode d'administration).
Inducteurs de la P-gp
Les données provenant d'une étude sur les interactions médicamenteuses chez des patients atteints de tumeurs solides à un stade avancé ont indiqué que l'administration concomitante d'une dose unique de 1 mg de talazoparib et de plusieurs doses quotidiennes d'un inducteur de la P-gp, la rifampicine à 600 mg, administrée 30 minutes avant la prise du talazoparib le jour même, a augmenté la Cmax du talazoparib de 37 % environ alors que l'AUCinf n'a pas été affectée, comparativement à une dose unique de 1 mg de talazoparib administrée seule. C'est probablement l'effet net de l'induction et de l'inhibition de la P-gp par la rifampicine dans les conditions de test de l'étude sur les interactions médicamenteuses. Aucune adaptation posologique du talazoparib n'est nécessaire lorsqu'il est co-administré avec la rifampicine. Toutefois, l'effet d'autres inducteurs de la P-gp sur l'exposition au talazoparib n'a pas été étudié. D'autres inducteurs de la P-gp (incluant mais ne se limitant pas à la carbamazépine, la phénytoïne et le millepertuis) peuvent diminuer l'exposition au talazoparib.
Inhibiteurs de la BCRP
L'effet des inhibiteurs de la BCRP sur la PK du talazoparib n'a pas été étudié in vivo. L'administration concomitante du talazoparib avec des inhibiteurs de la BCRP peut augmenter l'exposition au talazoparib. L'utilisation concomitante d'inhibiteurs puissants de la BCRP (incluant mais ne se limitant pas à curcumine et ciclosporine) doit être évitée. Si l'administration concomitante d'inhibiteurs puissants de la BCRP ne peut être évitée, le patient doit être surveillé afin de déceler tout effet indésirable accru potentiel.
Effet des antiacides
Une analyse de PK de population indique que l'administration concomitante d'antiacides incluant les inhibiteurs de la pompe à protons et les antagonistes des récepteurs à l'histamine de type 2 (H2RA) ou d'autres antiacides n'a pas d'impact significatif sur l'absorption du talazoparib.
Contraception hormonale systémique
Des études sur les interactions médicamenteuses entre le talazoparib et les contraceptifs n'ont pas été menées.
Le traitement par Talzenna doit être instauré et supervisé par un médecin expérimenté dans l'utilisation des médicaments anticancéreux.
Sélection des patients
Cancer du sein
Les patients doivent être sélectionnés pour le traitement du cancer du sein par Talzenna sur la base de la présence de mutations germinales BRCA délétères ou soupçonnées de l'être, déterminées par un laboratoire expérimenté utilisant une méthode de test validée.
Le conseil génétique pour les patients présentant des mutations BRCA doit être effectué, le cas échéant, conformément aux réglementations locales.
Cancer de la prostate
Il n'est pas nécessaire de procéder à une recherche mutationnelle pour sélectionner les patients atteints de CPRCm en vue d'un traitement par Talzenna.
Posologie
Talzenna en monothérapie (cancer du sein)
La dose recommandée est de 1 mg de talazoparib une fois par jour. Les patients doivent être traités jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
Talzenna en association avec l'enzalutamide (cancer de la prostate)
La dose recommandée est de 0,5 mg de talazoparib en association avec 160 mg d'enzalutamide une fois par jour. Les patients doivent être traités jusqu'à progression de la maladie ou survenue d'une toxicité inacceptable.
La castration médicale avec un analogue de l'hormone de libération de la lutéinostimuline (LHRH) doit être poursuivie pendant le traitement chez les patients n'ayant pas subi de castration chirurgicale.
Se référer au résumé des caractéristiques du produit de l'enzalutamide pour connaître la posologie recommandée.
Oubli de dose
En cas de vomissement ou d'oubli d'une dose de Talzenna, le patient ne doit pas prendre de dose supplémentaire. La dose prescrite suivante doit être prise à l'heure habituelle.
Adaptations posologiques
Pour la prise en charge des effets indésirables, une interruption du traitement ou une réduction posologique doit être envisagée selon la sévérité et la présentation clinique (voir tableau 1). Les paliers de réduction de doses recommandés pour le talazoparib en monothérapie (cancer du sein) et pour le talazoparib en association avec l'enzalutamide (cancer de la prostate) sont présentés dans le tableau 2 et le tableau 3, respectivement.
Un hémogramme doit être réalisé avant l'instauration du traitement par talazoparib et faire l'objet d'une surveillance mensuelle et si cliniquement indiqué (voir tableau 1 et rubrique Mises en garde spéciales et précautions d'emploi).
Tableau 1. Adaptations posologiques en cas d'effets indésirables
|
Suspendre le traitement par Talzenna jusqu'à ce que le taux revienne à |
Reprendre le traitement par Talzenna |
|
|
Hémoglobine < 8 g/dl |
≥ 9 g/dl |
Reprendre le traitement par Talzenna à la dose immédiatement inférieure |
|
Numération plaquettaire < 50 000/μl |
≥ 75 000/μl |
|
|
Numération des neutrophiles < 1 000/μl |
≥ 1 500/µl |
|
|
Effet indésirable non hématologique de grade 3 ou de grade 4 |
Grade ≤ 1 |
Envisager la reprise du traitement par Talzenna à la dose immédiatement inférieure ou l'arrêt du traitement |
Tableau 2. Paliers de réduction de doses pour le talazoparib en monothérapie (cancer du sein)
|
Palier de dose du talazoparib (cancer du sein) |
|
|
Dose initiale recommandée |
1 mg une fois par jour |
|
Première réduction posologique |
0,75 mg une fois par jour |
|
Deuxième réduction posologique |
0,5 mg une fois par jour |
|
Troisième réduction posologique |
0,25 mg une fois par jour |
Tableau 3. Paliers de réduction de doses pour le talazoparib en cas d'utilisation en association avec l'enzalutamide (cancer de la prostate)
|
Palier de dose du talazoparib (cancer de la prostate) |
|
|
Dose initiale recommandée |
0,5 mg une fois par jour |
|
Première réduction posologique |
0,35 mg une fois par jour |
|
Deuxième réduction posologique |
0,25 mg une fois par jour |
|
Troisième réduction posologique |
0,1 mg une fois par jour |
Se référer au résumé des caractéristiques du produit de l'enzalutamide pour l'adaptation posologique en cas d'effets indésirables associés à l'enzalutamide.
La gélule de 0,1 mg est destinée à faciliter les modifications posologiques et n'est pas interchangeable avec d'autres dosages.
Traitement concomitant par inhibiteurs de la glycoprotéine P (P-gp)
Talzenna en monothérapie (cancer du sein)
Les inhibiteurs puissants de la P-gp peuvent entraîner une augmentation de l'exposition au talazoparib. L'utilisation concomitante d'inhibiteurs puissants de la P-gp pendant le traitement par talazoparib doit être évitée. La co-administration ne doit être envisagée qu'après une évaluation attentive des bénéfices et des risques potentiels. Si la co-administration d'un inhibiteur puissant de la P-gp est inévitable, la dose de Talzenna doit être réduite à la dose inférieure suivante. Lors de l'arrêt de l'inhibiteur puissant de la P-gp, la dose de Talzenna doit être augmentée (après 3-5 demi-vies de l'inhibiteur de la P-gp) à la dose utilisée avant l'instauration de l'inhibiteur puissant de la P-gp (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Talzenna en association avec l'enzalutamide (cancer de la prostate)
L'effet de la co-administration d'inhibiteurs de la P-gp sur l'exposition au talazoparib lorsque le talazoparib est administré en association avec l'enzalutamide n'a pas été étudié. Par conséquent, l'utilisation concomitante d'inhibiteurs de la P-gp pendant le traitement par talazoparib doit être évitée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Populations particulières
Insuffisants hépatiques
Aucune adaptation posologique n'est nécessaire pour les patients présentant une insuffisance hépatique légère (bilirubine totale ≤ 1 × la limite supérieure de la normale [LSN] et aspartate aminotransférase [ASAT] > LSN, ou bilirubine totale > 1,0 à 1,5 × LSN et quelle que soit la valeur d'ASAT), modérée (bilirubine totale > 1,5 à 3,0 × LSN et quelle que soit la valeur d'ASAT) ou sévère (bilirubine totale > 3,0 × LSN et quelle que soit la valeur d'ASAT) (voir rubrique Propriétés pharmacocinétiques). L'utilisation de Talzenna en association avec l'enzalutamide n'est pas recommandée chez les patients présentant une insuffisance hépatique sévère (classe C sur l'échelle de Child-Pugh) car la pharmacocinétique et la sécurité n'ont pas été établies chez ces patients (voir rubrique Propriétés pharmacocinétiques).
Insuffisants rénaux
Cancer du sein
Aucune adaptation posologique n'est nécessaire pour les patients présentant une insuffisance rénale légère (60 ml/min ≤ clairance de la créatinine [ClCr] < 90 ml/min). Pour les patients présentant une insuffisance rénale modérée (30 ml/min ≤ ClCr < 60 ml/min), la dose initiale recommandée de
Talzenna est de 0,75 mg une fois par jour. Pour les patients présentant une insuffisance rénale sévère (15 ml/min ≤ ClCr < 30 ml/min), la dose initiale recommandée de Talzenna est de 0,5 mg une fois par jour. Talzenna n'a pas été étudié chez les patients présentant une ClCr < 15 ml/min ni chez les patients nécessitant une hémodialyse (voir rubrique Propriétés pharmacocinétiques).
Cancer de la prostate
Aucune adaptation posologique n'est nécessaire pour les patients présentant une insuffisance rénale légère (60 ml/min ≤ clairance de la créatinine [ClCr] < 90 ml/min). Pour les patients présentant une insuffisance rénale modérée (30 ml/min ≤ ClCr < 60 ml/min), la dose recommandée de Talzenna est de 0,35 mg une fois par jour en association avec l'enzalutamide une fois par jour par voie orale. Pour les patients présentant une insuffisance rénale sévère (15 ml/min ≤ ClCr < 30 ml/min), la dose recommandée de Talzenna est de 0,25 mg une fois par jour en association avec l'enzalutamide une fois par jour. Talzenna n'a pas été étudié chez les patients présentant une ClCr < 15 ml/min ni chez les patients nécessitant une hémodialyse (voir rubrique Propriétés pharmacocinétiques).
Sujets âgés
Aucune adaptation posologique n'est nécessaire chez les patients âgés (≥ 65 ans) (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité de Talzenna chez les enfants et adolescents âgés de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Talzenna doit être administré par voie orale. Pour éviter tout contact avec le contenu de la gélule, les gélules doivent être avalées entières et ne doivent être ni ouvertes ni dissoutes. Elles peuvent être prises avec ou sans nourriture (voir rubrique Propriétés pharmacocinétiques).
Durée de conservation :
4 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Il existe peu de cas de surdosage par le talazoparib. Aucun effet indésirable n'a été rapporté chez un patient s'étant autoadministré accidentellement trente gélules de 1 mg de talazoparib au Jour 1 et immédiatement traité par décontamination gastrique. Les symptômes de surdosage ne sont pas établis. En cas de surdosage, le traitement par talazoparib doit être arrêté et les médecins doivent envisager une décontamination gastrique, mettre en œuvre des soins de soutien généraux et traiter les symptômes.
Classe pharmacothérapeutique : antinéoplasiques, autres antinéoplasiques, Code ATC : L01XK04
Mécanisme d'action
Le talazoparib est un inhibiteur des enzymes PARP, PARP1 (CI50 = 0,7 nM) et PARP2 (CI50 = 0,3 nM). Les enzymes PARP sont impliquées dans les voies de signalisation de réponse aux lésions de l'ADN cellulaire, comme la réparation de l'ADN, la transcription génétique et la mort cellulaire. Les inhibiteurs de PARP (PARPi) exercent des effets cytotoxiques sur les cellules cancéreuses grâce à 2 mécanismes : l'inhibition de l'activité catalytique de PARP et le piégeage de PARP, par lequel la protéine PARP liée à un PARPi ne se dissocie pas facilement d'une lésion de l'ADN, ce qui empêche la réparation, la réplication et la transcription de l'ADN entrainant ainsi l'apoptose et/ou la mort cellulaire. Le traitement de lignées de cellules cancéreuses présentant des anomalies de gènes de réparation de l'ADN par le talazoparib en monothérapie entraîne une augmentation du taux de γH2AX, marqueur de cassures de l'ADN double brin et provoque une diminution de la prolifération cellulaire et une augmentation de l'apoptose. L'activité anti-tumorale du talazoparib a aussi été observée dans un modèle de xénogreffe du cancer du sein avec BRCA muté dérivée de patient (PDX) dans lequel le patient avait précédemment reçu un traitement à base de platine, ainsi que dans un modèle de xénogreffe du cancer de la prostate à récepteur aux androgènes (RA) positif. Dans ces modèles PDX, le talazoparib a réduit la croissance tumorale et a augmenté le taux de γH2AX et l'apoptose dans les tumeurs.
Les effets anti-tumoraux de l'inhibition combinée de l'activité PARP et RA reposent sur les mécanismes suivants : l'inhibition de la signalisation du RA supprime l'expression des gènes de réparation par recombinaison homologue (RRH), notamment BRCA1, ce qui entraîne une sensibilité à l'inhibition de PARP. Il a été démontré que l'activité de PARP1 était nécessaire à la fonction optimale du RA et que l'inhibition de PARP pouvait donc réduire la signalisation du RA et augmenter la sensibilité aux inhibiteurs de la signalisation du RA. La résistance clinique au blocage du RA est parfois associée à la codélétion de RB1 et BRCA2, qui est à son tour associée à la sensibilité à l'inhibition de PARP.
Électrophysiologie cardiaque
L'effet du talazoparib sur la repolarisation cardiaque a été évalué à l'aide d'électrocardiogrammes (ECG) appariés dans le temps pour évaluer la relation entre la modification de l'intervalle QT corrigé en fonction de la fréquence cardiaque (QTc) par rapport à l'inclusion, et les concentrations plasmatiques correspondantes de talazoparib chez 37 patients présentant des tumeurs solides à un stade avancé. Le talazoparib n'a pas eu d'effet cliniquement pertinent sur l'allongement de l'intervalle QTc à la dose cliniquement recommandée maximale en monothérapie de 1 mg une fois par jour.
Efficacité et sécurité cliniques
Cancer du sein localement avancé ou métastatique HER2 négatif avec mutations germinales BRCA (gBRCAm)
Étude EMBRACA
L'étude EMBRACA était une étude ouverte, randomisée, parallèle, à 2 bras, multicentrique comparant Talzenna à une chimiothérapie (capécitabine, éribuline, gemcitabine, vinorelbine) chez des patients atteints d'un cancer du sein localement avancé ou métastatique HER2 négatif avec mutations germinales BRCA, ayant reçu au maximum 3 protocoles de chimiothérapie cytotoxique antérieurs pour le traitement de leur cancer métastatique ou localement avancé. Les patients devaient avoir reçu une anthracycline et/ou un taxane (sauf contre-indication) dans le cadre d'un traitement néoadjuvant, adjuvant et/ou d'une maladie métastatique. Les patients ayant déjà reçu un traitement à base de platine contre un cancer avancé ne devaient présenter aucun signe de progression de la maladie pendant le traitement à base de platine. Aucun traitement antérieur par PARPi n'était autorisé.
Sur les 431 patients randomisés dans l'étude EMBRACA, 408 (95 %) ont reçu la confirmation par évaluation centrale d'une gBRCAm délétère ou soupçonnée d'être délétère à l'aide d'un test de l'étude clinique. Parmi ces patients, 354 (82 %) ont reçu la confirmation à l'aide du BRACAnalysis CDx. Le statut mutationnel du gène BRCA (cancer positif pour le gène de prédisposition au cancer du sein 1 [BRCA1] ou pour le gène de prédisposition au cancer du sein 2 [BRCA2]) était similaire dans les deux bras de traitement.
Au total, 431 patients ont été randomisés suivant un ratio de 2/1 pour recevoir Talzenna 1 mg, gélule, une fois par jour ou une chimiothérapie à des doses standard jusqu'à progression ou toxicité inacceptable. Sur les 431 patients randomisés dans l'étude EMBRACA, 287 ont été randomisés dans le bras Talzenna et 144 dans le bras chimiothérapie. La randomisation a été stratifiée selon le nombre de lignes antérieures de chimiothérapie pour le cancer métastatique (0 versus 1, 2 ou 3), selon le statut triple négatif du cancer (cancer du sein triple négatif [CSTN] versus non-[CSTN]) et selon les antécédents de métastases du système nerveux central (oui versus non).
Les données démographiques et caractéristiques médicales des patients à l'inclusion étaient généralement similaires entre les bras de traitement de l'étude (voir tableau 5).
Tableau 5. Données démographiques et caractéristiques médicales des patients à l'inclusion — Étude EMBRACA
|
|
Talazoparib (N = 287) |
Chimiothérapie (N = 144) |
|
Âge médian (ans [intervalle]) |
45,0 (27,0 ; 84,0) |
50,0 (24,0 ; 88,0) |
|
Catégorie d'âge (ans), n (%) |
||
|
< 50 |
182 (63,4 %) |
67 (46,5 %) |
|
50 à < 65 |
78 (27,2 %) |
67 (46,5 %) |
|
≥ 65 |
27 (9,4 %) |
10 (6,9 %) |
|
Sexe, n (%) |
||
|
Femmes |
283 (98,6 %) |
141 (97,9 %) |
|
Hommes |
4 (1,4 %) |
3 (2,1 %) |
|
Ethnie, n (%) |
||
|
Asiatiques |
31 (10,8 %) |
16 (11,1 %) |
|
Noirs ou Afro-américains |
12 (4,2 %) |
1 (0,7 %) |
|
Blancs |
192 (66,9 %) |
108 (75,0 %) |
|
Autre |
5 (1,7 %) |
1 (0,7 %) |
|
Non rapporté |
47 (16,4 %) |
18 (12,5 %) |
|
Indice de performance ECOG, n (%) |
||
|
0 |
153 (53,3 %) |
84 (58,3 %) |
|
1 |
127 (44,3 %) |
57 (39,6 %) |
|
2 |
6 (2,1 %) |
2 (1,4 %) |
|
Données manquantes |
1 (0,3 %) |
1 (0,7 %) |
|
Statut d'expression des récepteurs hormonaux, n (%) |
||
|
HER2 positif |
0 (0,0 %) |
0 (0,0 %) |
|
Triple-négatif |
130 (45,3 %) |
60 (41,7 %) |
|
Positif pour les récepteurs hormonaux (ER positif ou PgR positif) |
157 (54,7 %) |
84 (58,3 %) |
|
Statut du BRCA selon l'évaluation en laboratoire central ou local, n (%) |
287 (100,0 %) |
144 (100,0 %) |
|
Positif pour la mutation BRCA1 |
133 (46,3 %) |
63 (43,8 %) |
|
Positif pour la mutation BRCA2 |
154 (53,7 %) |
81 (56,3 %) |
|
Délai entre le diagnostic initial de cancer du sein et le diagnostic de cancer du sein avancé (années) |
||
|
n |
286 |
144 |
|
Médiane |
1,9 |
2,7 |
|
Minimum, maximum |
0 ; 22 |
0 ; 24 |
|
Catégories de délai entre le diagnostic initial de cancer du sein et le diagnostic de cancer du sein avancé |
||
|
< 12 mois |
108 (37,6 %) |
42 (29,2 %) |
|
≥ 12 mois |
178 (62,0 %) |
102 (70,8 %) |
|
Nombre de protocoles cytotoxiques antérieurs pour une maladie localement avancée ou métastatique |
||
|
Moyenne (écart type) |
0,9 (1,01) |
0,9 (0,89) |
|
Médiane |
1 |
1 |
|
Minimum, maximum |
0 ; 4 |
0 ; 3 |
|
Nombre de patients ayant déjà reçu un protocole cytotoxique pour une maladie localement avancée ou métastatique, n (%) |
||
|
0 |
111 (38,7 %) |
54 (37,5 %) |
|
1 |
107 (37,3 %) |
54 (37,5 %) |
|
2 |
57 (19,9 %) |
28 (19,4 %) |
|
3 |
11 (3,8 %) |
8 (5,6 %) |
|
≥ 4 |
1 (0,3 %) |
0 (0,0 %) |
Tableau 5. Données démographiques et caractéristiques médicales des patients à l'inclusion — Étude EMBRACA
|
|
Talazoparib (N = 287) |
Chimiothérapie (N = 144) |
|
Nombre de patients ayant reçu les traitements antérieurs suivants, n (%) |
|
|
|
Taxane |
262 (91,3 %) |
130 (90,3 %) |
|
Anthracycline |
243 (84,7 %) |
115 (79,9 %) |
|
Platine |
46 (16,0 %) |
30 (20,8 %) |
Abréviations : BRCA = gène de prédisposition au cancer du sein ; ER = récepteur aux œstrogènes ; HER2 = récepteur du facteur de croissance épidermique humain 2 ; N = nombre de patients ; n = nombre de patients dans la catégorie ; PgR = récepteur à la progestérone.
Le critère d'évaluation principal de l'efficacité était la survie sans progression (SSP) évaluée selon les critères d'évaluation de la réponse des tumeurs solides (RECIST), version 1.1, par une revue centralisée indépendante en aveugle (blindedindependent central review, BICR). Les objectifs secondaires étaient le taux de réponse objective (TRO), la survie globale (SG), la tolérance et la PK.
L'étude a démontré une amélioration statistiquement significative de la SSP, le critère d'évaluation principal, avec Talzenna par rapport à la chimiothérapie. Il n'y a pas eu d'effet statistiquement significatif sur la SG au moment de l'analyse finale de la SG. Les données d'efficacité de l'étude EMBRACA sont résumées dans le tableau 6. Les courbes de Kaplan-Meier pour la SSP et la SG sont présentées à la figure 1 et à la figure 3, respectivement.
Tableau 6. Résumé des résultats d'efficacité — Étude EMBRACA*
|
|
Talazoparib |
Chimiothérapie |
|
SSP par BICR |
N = 287 |
N = 144 |
|
Événements, nombre (%) |
186 (65 %) |
83 (58 %) |
|
Médiane (IC à 95 %), mois |
8,6 (7,2 ; 9,3) |
5,6 (4,2 ; 6,7) |
|
Rapport de risque a (IC à 95 %) |
0,54 (0,41 ; 0,71) |
|
|
Valeur de p bilatéraleb |
p < 0,0001 |
|
|
SG (analyse finale)c |
N = 287 |
N = 144 |
|
Événements, nombre (%) |
216 (75,3 %) |
108 (75 %) |
|
Médiane (IC à 95 %), mois |
19,3 (16,6 ; 22,5) |
19,5 (17,4 ; 22,4) |
|
Rapport de risque a (IC à 95 %) |
0,85 (0,67 ; 1,07)c |
|
|
Valeur de p bilatéraleb |
p = 0,1693 |
|
|
Réponse objective d'après l'investigateurd,e |
N = 219 |
N = 114 |
|
TRO, % (IC à 95 %) |
62,6 (55,8 ; 69,0) |
27,2 (19,3 ; 36,3) |
|
Odds ratio (IC à 95 %) |
4,99 (2,93 ; 8,83) |
|
|
Valeur de p bilatéralef |
p < 0,0001 |
|
|
Durée de réponse d'après l'investigateurd |
N = 137 |
N = 31 |
|
Médiane (IIQ), mois |
5,4 (2,8 ; 11,2) |
3,1 (2,4 ; 6,7) |
Abréviations : BICR = examen central indépendant en aveugle ; CMH = Cochran-Mantel-Haenszel ;
IC = intervalle de confiance ; IIQ = intervalle interquartile ; ITT = intention de traiter ; N = nombre de patients ; PARP = poly(adénosine diphosphate-ribose) polymérase ; RC = réponse complète ; RECIST 1.1 = critères d'évaluation de la réponse des tumeurs solides version 1.1 ; RP = réponse partielle ; SG = survie globale ; SSP = survie sans progression ; TRO = taux de réponse objective.
* La SSP, le TRO et la durée de réponse sont basés sur la date de gel des données du 15 septembre 2017 et un suivi médian pour la SSP de 13,0 mois (IC à 95 % : 11,1 ; 18,4) dans le bras talazoparib et de 7,2 mois (IC à 95 % : 4,6 ; 11,1) dans le bras chimiothérapie. La SG est basée sur la date de gel des données du 30 septembre 2019 et un suivi médian de 44,9 mois (IC à 95 % : 37,9 ; 47,0) dans le bras talazoparib et de 36,8 mois (IC à 95 % : 34,3 ; 43,0) dans le bras chimiothérapie.
a. Le rapport de risque était basé sur un modèle de régression de Cox stratifié, le traitement étant la seule covariable (facteurs de stratification : nombre de chimiothérapies cytotoxiques antérieures, statut triple négatif, antécédents de métastases au niveau du système nerveux central) et était relatif à la chimiothérapie globale, une valeur < 1 indiquant un résultat en faveur du talazoparib.
b. Test du log-rank stratifié.
c. Au moment de l'analyse finale de la SG, 46,3 % versus 41,7 % des patients randomisés dans les bras talazoparib et chimiothérapie, respectivement, ont reçu par la suite un traitement à base de platine, et 4,5 % versus 32,6 % ont reçu par la suite un traitement par inhibiteur de PARP.
d. Menée dans la population ITT avec maladie mesurable et réponse objective. Le taux de réponse complète a été de 5,5 % dans le bras talazoparib contre 0 % dans le bras chimiothérapie.
e. Selon les critères RECIST 1.1, confirmation de la RC/RP non nécessaire.
f. Test de CMH stratifié.
Figure 1. Courbes de Kaplan-Meier de la SSP — Étude EMBRAC
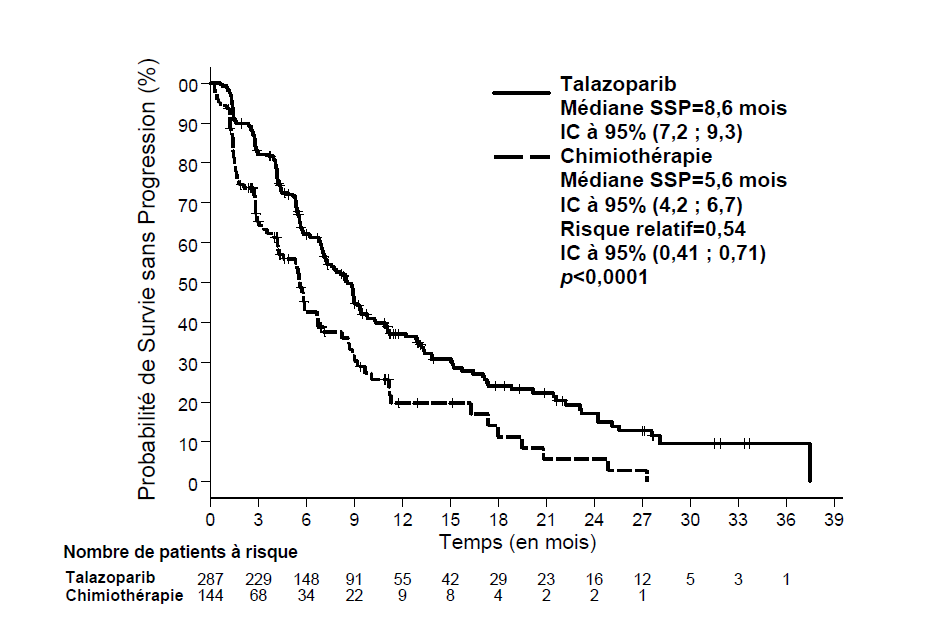
Abréviations : IC = intervalle de confiance ; SSP = survie sans progression.
Une série d'analyses de la SSP ont été effectuées dans des sous-groupes prédéfinis en fonction de facteurs pronostiques et de caractéristiques à l'inclusion afin d'évaluer la cohérence interne de l'effet du traitement. Conformément aux résultats globaux, une réduction du risque de progression de la maladie ou de décès en faveur du bras talazoparib a été observée dans tous les sous-groupes de patients individuels (figure 2).
Figure 2. Graphique en forêt des analyses de la SSP pour les sous-groupes principaux — Étude EMBRACA
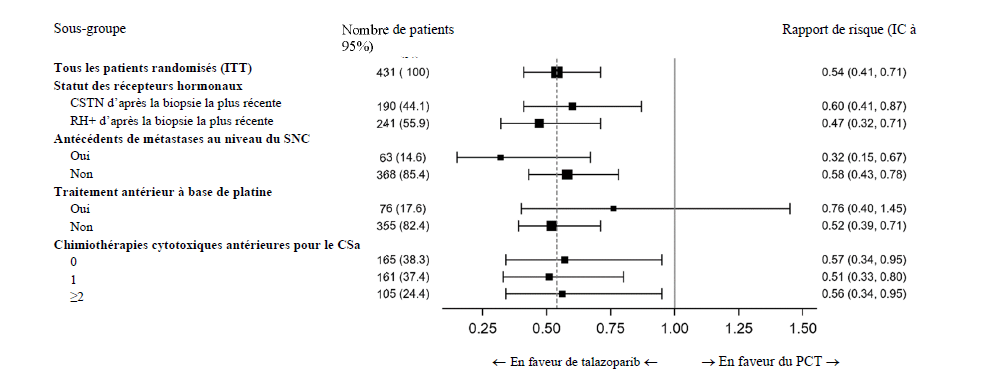
Abréviations : CSa = cancer du sein de stade avancé ; CSTN = cancer du sein triple négatif ; IC = intervalle de confiance ; ITT : intention de traiter ; PCT = traitement de choix du médecin (chimiothérapie) ; RH+ = positif pour les récepteurs hormonaux ; SNC = système nerveux central ; SSP = survie sans progression.
Figure 3 Courbes de Kaplan-Meier de la survie globale — Étude EMBRACA
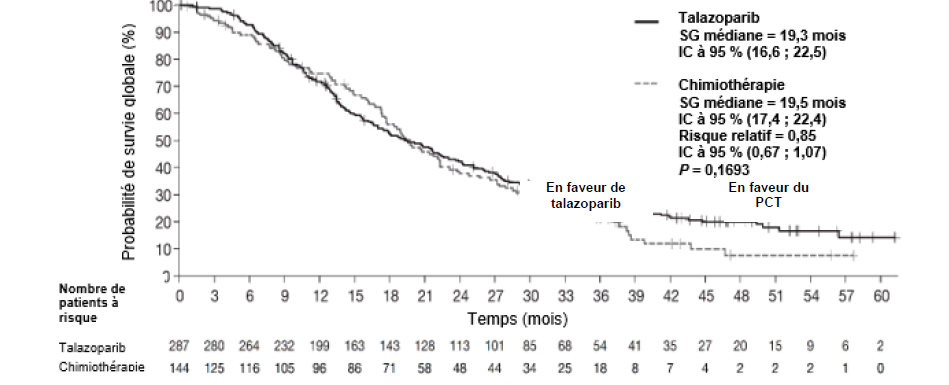
Abréviations : IC = intervalle de confiance ; SG = survie globale.
La valeur de p de l'analyse principale était basée sur un test du log-rank stratifié.
Cancer de la prostate métastatique résistant à la castration (CPRCm)
Étude TALAPRO-2
L'étude TALAPRO-2 était une étude randomisée, en double aveugle, contrôlée contre placebo, au cours de laquelle des patients (N = 805) atteints de CPRCm ont été randomisés selon un rapport de 1:1 pour recevoir Talzenna 0,5 mg une fois par jour en association avec l'enzalutamide 160 mg une fois par jour, versus un bras comparateur recevant un placebo en association avec l'enzalutamide 160 mg une fois par jour. Tous les patients avaient reçu un analogue de l'hormone de libération de la gonadotrophine (GnRH) ou avaient subi une orchidectomie bilatérale antérieure et ils devaient avoir progressé lors d'un traitement antérieur par déprivation androgénique. Un traitement antérieur par abiratérone ou chimiothérapie à base de taxane pour un cancer de la prostate métastatique sensible à la castration (CPSCm) était autorisé.
La randomisation a été stratifiée en fonction (1) du traitement antérieur par abiratérone ou chimiothérapie à base de taxane versus l'absence d'un tel traitement antérieur ; et (2) du statut d'altération des gènes de RRH qui a été testé prospectivement par séquençage de nouvelle génération du tissu tumoral à l'aide du FoundationOneCDx ou de l'ADN tumoral circulant (ADNtc) à l'aide du FoundationOneLiquidCDx ; les patients présentant des altérations des gènes de RRH (ATM, ATR, BRCA1, BRCA2, CDK12, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2 ou RAD51C) versus les patients ne présentant pas d'altérations des gènes de RRH ou dont le statut n'était pas connu.
L'âge médian était de 71 ans (intervalle de 36 à 91 ans) dans les deux bras ; 62 % des patients étaient blancs, 31 % asiatiques et 2 % noirs. La plupart des participants (66 %) dans les deux bras avaient un statut de performance ECOG de 0. Chez les patients traités par Talzenna la proportion de patients présentant une maladie mesurable à l'inclusion selon les critères RECIST 1.1, par revue centralisée indépendante en aveugle (blindedindependent central review, BICR), était de 30 %. Vingt-huit pour cent (28 %) des patients avaient reçu un traitement antérieur par abiratérone ou une chimiothérapie à base de taxane. Vingt pour cent (20 %) avaient des tumeurs présentant des altérations des gènes de RRH et 80 % des tumeurs ne présentant pas d'altération des gènes de RRH ou avaient un statut inconnu.
Le critère d'efficacité principal était la survie sans progression radiographique (SSPr) évaluée selon les critères RECIST version 1.1 et Prostate Cancer Clinical Trials Working Group Criteria 3 (PCWG3) (os), tels qu'évalués par BICR. La SG était un critère d'évaluation secondaire avec contrôle du risque alpha.
Une amélioration statistiquement significative de la SSPr évaluée par une revue centralisée indépendante en aveugle (BICR) a été démontrée pour Talzenna en association avec l'enzalutamide par rapport au placebo en association avec l'enzalutamide. Une analyse de sensibilité de la SSPr évaluée par l'investigateur a été cohérente avec les résultats de la SSPr évaluée par la revue centralisée indépendante en aveugle (BICR).
Les résultats d'efficacité de l'étude TALAPRO-2 sont présentés dans le tableau 7 et la figure 4.
Tableau 7. Résumé des résultats d'efficacité — TALAPRO-2 (CPRCm)*
|
|
Talazoparib + enzalutamide |
Placebo + enzalutamide |
|
SSPr par BICR |
N = 402 |
N = 403 |
|
Événements, nombre (%) |
151 (37,6) |
191 (47,4) |
|
Médiane en mois (IC à 95 %) |
NA (27,5 ; NA) |
21,9 (16,6 ; 25,1) |
|
Rapport de risque (IC à 95 %)a Valeur de pb |
0,627 (0,506 ; 0,777) p < 0,0001 |
|
|
Deuxième analyse intermédiaire SG |
|
|
|
Événements, nombre (%) |
156 (38,8) |
174 (43,2) |
|
Médiane en mois (IC à 95 %) |
NA (37,3 ; NA) |
38,2 (34,1 ; 43,1) |
|
Rapport de risque (IC à 95 %)a |
0,837 (0,674 ; 1,040) |
|
Abréviations : BICR = revue centralisée indépendante en aveugle ; IC = intervalle de confiance ;
CPSC = cancer de la prostate sensible à la castration ; RRH = réparation par recombinaison homologue ;
CPRCm = cancer de la prostate métastatique résistant à la castration ; N = nombre de patients ;
NHT = hormonothérapie de nouvelle génération ; NA = non atteint ; SG = survie globale ; SSPr = survie sans progression radiographique.
* La SSPr est basée sur la date de gel des données du 16 août 2022 et un suivi médian pour la SSPr de 24,9 mois (IC à 95 % : 24,7 ; 25,3) dans le bras talazoparib plus enzalutamide et de 24,6 mois (IC à 95 % : 22,1 ; 24,9) dans le bras placebo plus enzalutamide. La deuxième analyse intermédiaire de SG est basée sur la date de gel des données du 28 mars 2023 et un suivi médian de 35,8 mois (IC à 95 % : 33,6 ; 35,9) dans le bras talazoparib plus enzalutamide et de 34,6 mois (IC à 95 % : 32,7 ; 35,9) dans le bras placebo plus enzalutamide.
a Rapport de risque basé sur le modèle des
risques proportionnels de Cox, stratifié en fonction du traitement antérieur
par NHT (abiratérone) ou chimiothérapie à base de
taxane pour le CPSC (oui versus non) et en fonction du statut
mutationnel RRH (déficit versus aucun déficit/non connu), une
valeur < 1 indiquant un résultat en faveur du talazoparib.
b. Valeurs de p (bilatérales)
du test du log-rank stratifié en fonction du
traitement antérieur par NHT (abiratérone) ou par
chimiothérapie à base de taxane pour le CPSC et en fonction du statut
mutationnel RRH
Tableau 8. Résumé des résultats d'efficacité pour l'analyse en sous-groupes —
Étude TALAPRO-2 (CPRCm)*
|
|
Talazoparib + enzalutamide |
Placebo + enzalutamide |
|
Analyses de sous-groupes RRHma |
||
|
RRHm |
N = 85 |
N = 82 |
|
SSPr par BICR |
|
|
|
Événements, nombre (%) |
37 (43,5) |
49 (59,7) |
|
Médiane en mois (IC à 95 %) |
27,9 (16,8 ; NA) |
13,8 (10,9 ; 19,5) |
|
Rapport de risque (IC à 95 %)b |
0,424 (0,275 ; 0,653) |
|
|
Deuxième analyse intermédiaire SG |
|
|
|
Événements, nombre (%) |
30 (35,3) |
41 (50,0) |
|
Médiane en mois (IC à 95 %) |
41,9 (36,4 ; NA) |
30,8 (25,6 ; 38,8) |
|
Rapport de risque (IC à 95 %)b |
0,516 (0,320 ; 0,831) |
|
|
Non RRHm |
N = 207 |
N = 219 |
|
SSPr par BICR |
|
|
|
Événements, nombre (%) |
73 (35,3) |
95 (43,4) |
|
Médiane en mois (IC à 95 %) |
NA (25,8 ; NA) |
22,4 (16,6 ; NA) |
|
Rapport de risque (IC à 95 %)b |
0,695 (0,511 ; 0,944) |
|
|
Deuxième analyse intermédiaire SG |
|
|
|
Événements, nombre (%) |
82 (39,6) |
96 (43,8) |
|
Médiane en mois (IC à 95 %) |
NA (33 ; NA) |
38 (33,9 ; NA) |
|
Rapport de risque (IC à 95 %)b |
0,880 (0,654 ; 1,182) |
|
|
Analyses de sous-groupes BRCAma |
||
|
BRCAm |
N = 27 |
N = 32 |
|
SSPr par BICR |
|
|
|
Événements, nombre (%) |
8 (29,6) |
22 (68,7) |
|
Médiane en mois (IC à 95 %) |
NA (16.8 ; NA) |
11 (7,4 ; 24,6) |
|
Rapport de risque (IC à 95 %)b |
0.232 (0.101 ; 0.529) |
|
|
Deuxième analyse intermédiaire SG |
|
|
|
Événements, nombre (%) |
12 (44,4) |
18 (56,3) |
|
Médiane en mois (IC à 95 %) |
41,9 (24,9 ; NA) |
26,1 (15,2 ; NA) |
|
Rapport de risque (IC à 95 %)b |
0,558 (0,263 ; 1,187) |
|
Abréviations : BICR = examen central indépendant en aveugle ; BRCAm = gène du cancer du sein muté
(BReast Cancer, BRCA) ; IC = intervalle de confiance ; CPSC = cancer de la prostate sensible à la castration ;
ADNtc = ADN tumoral circulant ; RRHm = gène de réparation par recombinaison homologue muté ;
CPRCm = cancer de la prostate métastatique résistant à la
castration ; N = nombre de patients ;
NHT = hormonothérapie
de nouvelle génération ; NA = non atteint ; SG = survie globale ; SSPr = survie sans progression radiographique.
* Sur la base de la
date de gel des données du 16 août 2022 et d'un suivi médian de la SSPr de 24,9 mois (IC à 95 % : 24,7 ; 25,3) dans le bras talazoparib plus enzalutamide et
de 24,6 mois (IC à 95 % : 22,1 ; 24,9) dans le bras placebo plus enzalutamide. La deuxième analyse intermédiaire de SG est
basée sur la date de gel des données du 28 mars 2023 et sur un suivi médian de
35,8 mois (IC à 95 % : 33,6 ; 35,9) dans le bras talazoparib
plus enzalutamide et de 34,6 mois (IC à 95 % : 32,7 ;
35,9) dans le bras placebo plus enzalutamide.
a. Calculées sur la base des résultats prospectifs des tissus tumoraux (résultats connus avant la randomisation) et des résultats prospectifs de l'ADNtc dans le sang (résultats connus avant la randomisation).
b Rapport de risque basé sur le modèle des risques proportionnels de Cox, stratifié en fonction du traitement antérieur par NHT (abiratérone) ou chimiothérapie à base de taxane pour le CPSC (oui versus non), une valeur < 1 indiquant un résultat en faveur du talazoparib.
Figure 4. Courbes de Kaplan-Meier de la SSPr par BICR — Étude TALAPRO-2 (CPRCm)
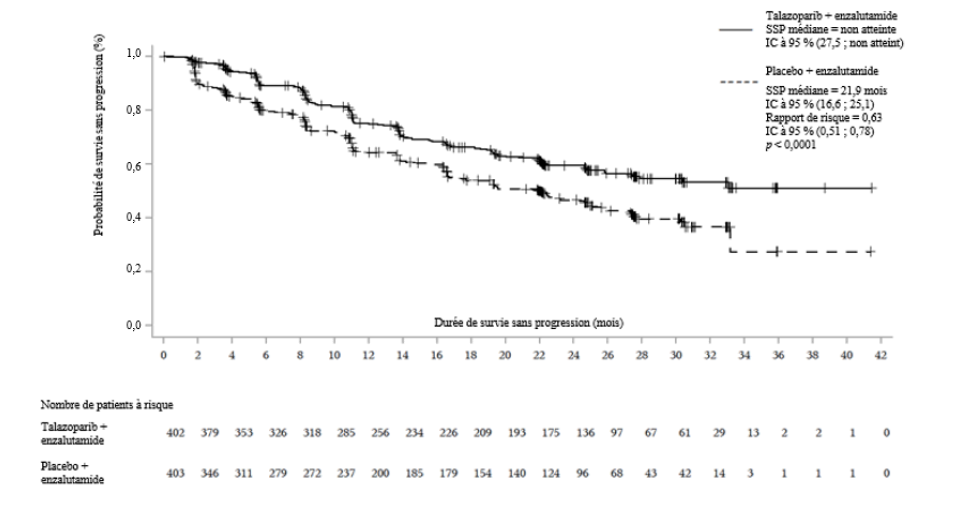
Abréviations : BICR = revue centralisée
indépendante en aveugle ; IC = intervalle de confiance ;
CPRCm
= cancer de la prostate métastatique résistant à la castration ;
SSP = survie sans progression ; SSPr = survie sans progression radiographique.
Figure 5. Diagramme en forêt des analyses de SSPr pour les principaux sous-groupes prédéfinis — Étude TALAPRO-2 (CPRCm)
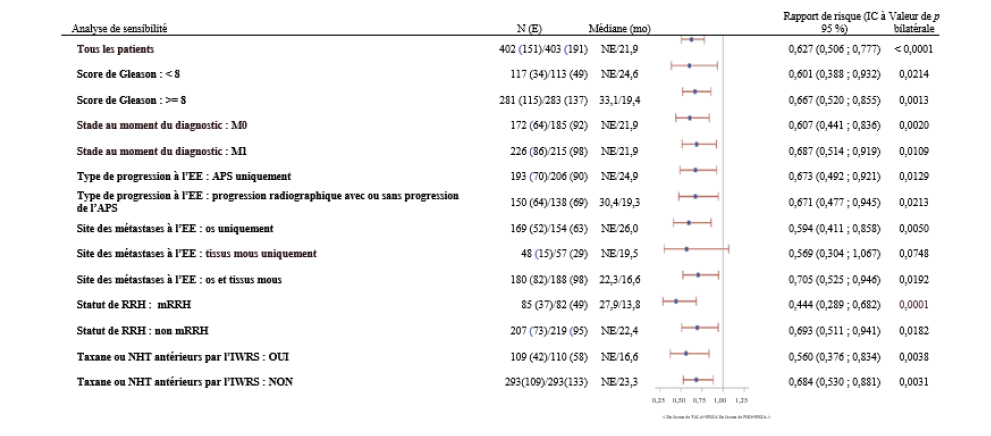
Abréviations : IC = intervalle de confiance ; ADNtc = ADN tumoral circulant ; ENZA = enzalutamide ; RRH = réparation par recombinaison homologue ; RRHm = gène de réparation par recombinaison homologue muté ; IWRS = Interactive Web Response System (système de réponse Web interactif) ; CPRCm = cancer de la prostate métastatique résistant à la castration ; N = nombre de participants ; NE = non évaluable/non atteint ; NHT = hormonothérapie de nouvelle génération ; PBO = placebo ; APS = antigène prostatique spécifique ; SSPr = survie sans progression radiographique ; EE = entrée dans l'étude ; TALA = talazoparib.
Le rapport de risque pour tous les patients était basé sur un modèle de Cox stratifié par les facteurs de stratification de randomisation. Pour tous les sous-groupes, le rapport de risque était basé sur un modèle de Cox non stratifié avec le traitement comme seule covariable. Un rapport de risque < 1 favorise le talazoparib. Le statut RRH est dérivé sur la base de résultats prospectifs basés sur les tissus tumoraux et de résultats prospectifs d'ADNc sanguins.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec le talazoparib dans tous les sous-groupes de la population pédiatrique, dans l'indication du cancer du sein et du cancer de la prostate (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
L'exposition au talazoparib a augmenté proportionnellement à la dose pour des doses comprises entre 0,025 mg et 2 mg après administration quotidienne de doses répétées. Après administrations quotidiennes répétées de 1 mg de talazoparib en monothérapie aux patients atteints d'un cancer du sein, la moyenne géométrique (coefficient de variation en % [CV %]) de l'aire sous la courbe de concentration plasmatique en fonction du temps (AUC) et la concentration plasmatique maximale observée (Cmax) du talazoparib à l'état d'équilibre étaient incluses entre 126 (107) ng•h/ml et 208 (37) ng•h/ml et entre 11 (90) ng/ml et 19 (27) ng/ml, respectivement. Après administration orale de 0,5 mg de talazoparib une fois par jour en association avec l'enzalutamide chez des patients atteints de CPRCm, la moyenne géométrique (CV %) de la Crésiduelle à l'état d'équilibre au cours des visites était comprise entre 3,29 et 3,68 ng/ml (45 à 48 %), ce qui était similaire aux valeurs observées de 3,53 ng/ml (61 %) lorsque le talazoparib en monothérapie était administré à la dose de 1 mg une fois par jour chez des patients atteints d'un cancer du sein. Après administrations quotidiennes répétées, les concentrations plasmatiques de talazoparib ont atteint l'état d'équilibre en 2 à 3 semaines en cas d'administration en monothérapie, et en 9 semaines environ en cas de co-administration avec l'enzalutamide. Le rapport d'accumulation médian de talazoparib après administrations par voie orale répétées de 1 mg en monothérapie une fois par jour était compris entre 2,3 et 5,2. Le talazoparib est un substrat des transporteurs de la P-gp et de la BCRP.
Absorption
Après administration orale de talazoparib, le temps médian jusqu'à la Cmax (Tmax) a été généralement compris entre 1 et 2 heures. L'étude de biodisponibilité absolue n'a pas été menée chez l'Homme. Cependant, d'après les données sur l'excrétion urinaire, la biodisponibilité absolue est d'au moins 41 %, avec une fraction absorbée d'au moins 69 % (voir Élimination). On ne s'attend pas à ce que les antiacides aient un effet significatif sur l'exposition au talazoparib, étant donné que la solubilité du talazoparib est suffisante à tous les pH entre 1 et 6,8. Vingt-huit pour cent (28 %) des patients de l'étude pivot prenaient des antiacides, principalement des inhibiteurs de la pompe à protons.
Effet de la nourriture
La prise de nourriture réduit la vitesse mais pas l'ampleur de l'absorption du talazoparib. Après une seule dose par voie orale de talazoparib prise avec un repas à forte teneur en lipides et en calories (environ 827 calories, 57 % de lipides), la Cmax moyenne du talazoparib a diminué d'environ 46 %, le Tmax médian a été allongé de 1 à 4 heures, tandis que l'AUCinf n'a pas été affectée. Compte tenu de ces résultats, Talzenna peut être administré avec ou sans nourriture (voir rubrique Posologie et mode d'administration).
Distribution
Le volume de distribution apparent moyen (Vss/F) du talazoparib dans la population était de 420 l. In vitro, la liaison du talazoparib aux protéines plasmatiques est d'environ 74 % et elle est indépendante de la concentration sur l'intervalle de concentration de 0,01 µM à 1 µM. L'insuffisance rénale ou hépatique ne semble pas avoir d'impact sur la liaison du talazoparib aux protéines, car aucune tendance évidente n'a pu être observée au niveau de la fraction moyenne non liée du talazoparib (fu) dans le plasma humain in vivo en cas de détérioration de la fonction rénale ou de la fonction hépatique.
Biotransformation
Le talazoparib subit un métabolisme hépatique minime chez l'Homme. Après administration orale d'une dose unique de 1 mg de talazoparib marqué au carbone 14 chez l'Homme, aucun métabolite circulant majeur n'a été identifié dans le plasma, et le talazoparib a été la seule entité dérivée du médicament identifiée dans la circulation. Aucun métabolite représentant individuellement plus de 10 % de la dose administrée n'a été récupéré dans l'urine ni dans les fèces.
In vitro, le talazoparib n'est pas un inhibiteur du cytochrome (CYP)1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4/5 ni un inducteur de CYP1A2, CYP2B6 ou CYP3A4 aux concentrations cliniquement pertinentes.
In vitro, le talazoparib n'a inhibé aucun des principaux transporteurs membranaires intestinaux, hépatiques ou rénaux (P-gp, BCRP, polypeptide de transport d'anions organiques [OATP]1B1,
OATP1B3, transporteur de cations organiques [OCT]1, OCT2, transporteur d'anions organiques [OAT]1, OAT3, pompe d'exportation des sels biliaires [BSEP], extrusion de multiples médicaments et toxines [MATE]1 et MATE2-K) aux concentrations cliniquement pertinentes.
In vitro, le talazoparib n'a inhibé aucune des principales isoformes de l'uridine-diphosphate glucuronosyltransférase (UGT) (1A1, 1A4, 1A6, 1A9, 2B7 et 2B15) aux concentrations cliniquement pertinentes.
Élimination
L'élimination rénale du médicament inchangé (filtration passive et sécrétion active) constitue la principale voie d'élimination du talazoparib. La P-gp est probablement impliquée dans la sécrétion rénale active du talazoparib. La demi-vie plasmatique terminale moyenne (± écart type) du talazoparib a été de 90 (± 58) heures et la clairance (CL/F) apparente moyenne de la population (variabilité interindividuelle) par voie orale de 6,5 (31 %) l/h chez les patients atteints de cancer. Chez 6 femmes ayant reçu une dose orale unique de talazoparib marqué au carbone 14, en moyenne 69 % (± 8,6 %) et 20 % (± 5,5 %) de la dose radioactive totale administrée a été récupérée dans l'urine et les fèces, respectivement. L'excrétion du talazoparib inchangé dans l'urine était la voie d'élimination principale, représentant 55 % de la dose administrée ; 14 % du talazoparib a été retrouvé dans les fèces sous forme inchangée.
Populations particulières
Âge, sexe et poids corporel
Une analyse de PK de population a été réalisée en utilisant les données de 490 patients atteints de cancer ayant reçu 1 mg de talazoparib par jour en monothérapie, afin d'évaluer l'impact de l'âge (de 18 à 88 ans), du sexe (53 hommes et 437 femmes) et du poids corporel (allant de 35,7 kg à 162 kg) sur la PK du talazoparib. Les résultats ont montré que l'âge, le sexe et le poids corporel n'avaient aucun effet clinique pertinent sur la PK du talazoparib.
Origine ethnique
D'après une analyse de PK de population portant sur 490 patients ayant reçu 1 mg de talazoparib par jour en monothérapie, dont 41 étaient asiatiques et 449 non asiatiques (361 Blancs, 16 Noirs, 9 autres et 63 non rapportés), la CL/F du talazoparib était plus élevée chez les patients asiatiques que chez les patients non asiatiques, entrainant une exposition (AUC) inférieure de 19 % chez les patients asiatiques.
Population pédiatrique
La pharmacocinétique du talazoparib n'a pas été évaluée chez les patients âgés de moins de 18 ans.
Insuffisants rénaux
Talazoparib en monothérapie
Les données issues d'une étude PK menée chez des patients atteints d'un cancer avancé et présentant divers degrés d'insuffisance rénale ont indiqué que l'exposition totale (AUC0-24) au talazoparib après l'administration de plusieurs doses quotidiennes de talazoparib a augmenté de 92 % et de 169 % chez les patients atteints d'insuffisance rénale modérée (DFGe 30 - < 60 ml/min) et sévère (DFGe < 30 ml/min), respectivement, par rapport aux patients ayant une fonction rénale normale (DFGe ≥ 90 ml/min). La Cmax du talazoparib a augmenté de 90 % et 107 % chez les patients atteints d'insuffisance rénale modérée et sévère, respectivement, par rapport aux patients ayant une fonction rénale normale. L'exposition au talazoparib a été similaire chez les patients atteints d'insuffisance rénale légère (DFGe 60 - < 90 ml/min) et chez ceux ayant une fonction rénale normale. En outre, d'après une analyse de PK de population incluant 490 patients, dont 132 patients atteints d'insuffisance rénale légère (60 ml/min ≤ ClCr < 90 ml/min), 33 patients atteints d'une insuffisance rénale modérée (30 ml/min ≤ ClCr < 60 ml/min), et 1 patient atteint d'une insuffisance rénale sévère (ClCr < 30 ml/min), la CL/F du talazoparib a diminué de 14 % et de 37 % chez les patients atteints d'insuffisance rénale légère et modérée, correspondant à une augmentation de 17 % et de 59 % de l'AUC, respectivement, par rapport aux patients ayant une fonction rénale normale (ClCr ≥ 90 ml/min). La PK du talazoparib n'a pas été étudiée chez les patients nécessitant une hémodialyse (voir rubrique Posologie et mode d'administration).
Talazoparib co-administré avec l'enzalutamide
D'après une analyse PK de population incluant 412
patients atteints de CPRCm ayant reçu du talazoparib co-administré avec
l'enzalutamide, dont 152 patients atteints d'une insuffisance rénale légère (60
ml/min ≤ ClCr < 90 ml/min), 72 patients atteints d'une insuffisance rénale
modérée (30 ml/min ≤ ClCr < 60 ml/min), et 2 patients atteints d'une
insuffisance rénale sévère (ClCr < 30 ml/min), la CL/F du talazoparib a
diminué de 8% et 27 %, correspondant à une augmentation de 9 % et 37 % de
l'AUC, chez les patients présentant respectivement une insuffisance rénale
légère à modérée, par rapport aux patients ayant une fonction rénale normale.
La PK du talazoparib n'a pas été étudiée chez les patients nécessitant une
hémodialyse (voir rubrique Posologie et mode d'administration).
Insuffisants hépatiques
Talazoparib en monothérapie
D’après une analyse PK de population incluant 490 patients ayant reçu 1 mg de talazoparib par jour en monothérapie, dont 118 patients atteints d’insuffisance hépatique légère (bilirubine totale ≤ 1,0 × LSN et ASAT > LSN, ou bilirubine totale > 1,0 à 1,5 × LSN et quelle que soit la valeur d’ASAT), l’insuffisance hépatique légère n’a pas eu d’effet sur la PK du talazoparib. La PK du talazoparib chez les patients présentant une fonction hépatique normale, une insuffisance hépatique légère, une insuffisance hépatique modérée (bilirubine totale > 1,5 à 3,0 × LSN et quelle que soit la valeur d’ASAT) ou une insuffisance hépatique sévère (bilirubine totale > 3,0 × LSN et quelle que soit la valeur d’ASAT) a été étudiée dans une étude de PK. Une analyse de population réalisée à partir des données de cette étude PK a indiqué que l’insuffisance hépatique légère, modérée ou sévère n’avait pas d’impact significatif sur la PK du talazoparib (voir rubrique Posologie et mode d'administration).
Talazoparib co-administré avec l'enzalutamide
La PK du talazoparib en association avec l'enzalutamide n'a pas été étudiée chez les patients présentant une insuffisance hépatique (voir rubrique Posologie et mode d'administration).
Talzenna a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. L'administration du talazoparib peut entraîner une fatigue/asthénie ou des sensations vertigineuses.
Lorsque Talzenna est administré en association avec l'enzalutamide, consulter le résumé des caractéristiques du produit de l'enzalutamide pour connaître les effets de l'enzalutamide sur l'aptitude à conduire des véhicules et à utiliser des machines.
Cancérogenèse
Aucune étude de cancérogenèse n'a été menée avec le talazoparib.
Génotoxicité
Le talazoparib ne s'est pas révélé mutagène dans un test de mutation inverse sur bactéries (test d'Ames). Le talazoparib s'est avéré clastogène dans un test d'aberrations chromosomiques in vitro sur lymphocytes du sang périphérique humain et dans un test des micronoyaux in vivo chez le rat, à des expositions similaires à celles obtenues avec des doses cliniquement pertinentes. Cette clastogénicité est cohérente avec l'instabilité génomique résultant de la pharmacologie primaire du talazoparib et indique un risque potentiel de génotoxicité chez l'Homme.
Toxicité en administration répétée
Dans des études de toxicité en administration répétée chez le rat et le chien, les principaux résultats à des expositions subthérapeutiques ont inclus une hypocellularité de la moelle osseuse avec une diminution dose-dépendante des cellules hématopoïétiques, une déplétion du tissu lymphoïde dans plusieurs organes et une atrophie et/ou des modifications dégénératives des testicules, de l'épididyme et des tubules séminifères. Les observations supplémentaires à des expositions supérieures ont inclus une augmentation dose-dépendante de l'apoptose/nécrose dans le tractus gastro-intestinal (GI), le foie et les ovaires. La plupart des observations histopathologiques étaient généralement réversibles et les observations au niveau des testicules étaient partiellement réversibles après 4 semaines sans traitement. Ces résultats de toxicité sont en accord avec la pharmacologie du talazoparib et avec sa distribution tissulaire.
Toxicologie du développement
Dans une étude du développement embryo-fœtal menée chez des rats, le talazoparib a entraîné des décès embryo-fœtaux, des malformations fœtales (enfoncement des yeux, microphtalmie, sternèbres fendues, arc vertébral cervical fusionné) et des variations structurelles osseuses à une exposition maternelle systémique AUC24 correspondant à environ 0,09 fois l'exposition correspondante chez l'Homme à la dose recommandée.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament
nécessitant une surveillance particulière pendant le traitement.
Prescription hospitalière.
Prescription réservée aux médecins compétents en CANCEROLOGIE.
Prescription réservée aux spécialistes
et services ONCOLOGIE MEDICALE.
Gélule.
Talzenna 0,25 mg, gélules
Gélule opaque, d'environ 14 mm × 5 mm, avec une coiffe ivoire (comportant la mention « Pfizer » imprimée en noir) et un corps blanc (comportant la mention « TLZ 0.25 » imprimée en noir).
Flacon en polyéthylène haute densité (PEHD) muni d'un bouchon en polypropylène (PP) à revêtement pour scellage par induction thermique.
Conditionnement : boîte de 30 gélules contenues dans un flacon en PEHD.
Chaque gélule contient du tosylate de talazoparib équivalant à 0,25 mg de talazoparib.
Contenu de la gélule
Cellulose microcristalline silicifiée (cellulose microcristalline et dioxyde de silicium)
Enveloppe de la gélule de 0,25 mg
Hypromellose
Oxyde de fer jaune (E172)
Dioxyde de titane (E171)
Encre d'impression
Gomme laque (E904)
Propylène glycol (E1520)
Hydroxyde d'ammonium (E527)
Oxyde de fer noir (E172)
Hydroxyde de potassium (E525)